





 Research Teams
Research Teams

Team Arima
Elucidation of the mechanisms underlying human placental development and design of a placenta-on-a-chip platform
Takahiro Arima:Tohoku University, Department Graduate School of Medicine, Professor
Hirokazu Kaji:Tohoku University, Graduate School of Engineering, Associate Professor
Mikita Suyama:Kyushu University Medical Institute of Bioregulation, Professor
Overview
The placenta is a multifunctional organ that provides nutrients to the fetus and taking away waste, produces hormones to maintain pregnancy, and protects the fetus from the maternal immune system. Poor placentation is associated with many diseases such as hypertensive disorders of pregnancy (HDP), small for gestational age (SGA), miscarriage, preterm birth, and hydatidiform moles. This study aims to determine the epigenomes of diseased placentae, clarify disease-specific epigenetic changes, and identify diagnostic biomarkers. To this end, we will adapt and optimize cutting-edge epigenetic profiling techniques.
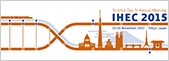
Previously, we generated and released the standard epigenome profiles of the constituent cells of the human placenta, which satisfy the strict IHEC criteria for DNA methylation, histone modifications, and small RNA. Combined with DNA methylation profiles of human oocytes, sperm, and blastocysts, we have uncovered the unique DNA methylation dynamics during early human development: (1) lower methylation levels than embryonic tissues and large partially methylated domains (PMDs) covering ~40% of the genome; (2) much less demethylation in the oocyte genome in the human blastocyst and carry-over to the placenta; (3) many placenta-specific differentially methylated regions; (4) skewed X-chromosome inactivation (XCI) and identification of X-linked imprinted genes (PLoS Genet. 2014, Am J Hum Genet. 2016). These contents were introduced as an important outcome of IHEC in Cell (Stunnenberd et al. Cell 2016).
We also succeeded in the long-term culture of human trophoblast stem (TS) cells derived from CT cells and blastocysts for the first time in the world (Cell Stem Cell 2018). These cells can be stably maintained in an undifferentiated state for more than 5 months without losing their ability to differentiate into two major trophoblast subtypes, syncytiotrophoblast (ST) and extravillous cytotrophoblast (EVT). ST cells are multinucleated cells formed by fusion and produce large quantities of placental hormones. EVT cells invade the decidualized endometrium and remodel the spiral arteries. Importantly, human TS cells and their differentiated derivatives had gene expression and DNA methylation patterns very similar to primary trophoblast cells. Moreover, when injected into NOD/SCID immunodeficient mice, human TS cells gave rise to ST-like cells containing blood-filled lacunae, reminiscent of primitive ST cells that form during implantation, invade the maternal endometrium, and erode maternal sinusoids. Also, the host mouse serum contained a large amount of human chorionic gonadotropin (hCG). Therefore, human TS cells will be a powerful tool to understand human placental development and functions.
In this study, we aim to understand the molecular mechanisms underlying human placental development and pregnancy-related diseases. 1) We perform epigenome analyses of placentas obtained from HDP and SGA patients and identify epigenetic mutations for a better understanding of the etiology and pathophysiology of these diseases. We also identify diagnostic biomarkers using samples from a large cohort study. 2) We establish disease-specific TS cells from HDP and SGA patients and clarify the underlying molecular mechanisms. 3) We register "disease epigenome" obtained from the placental samples and disease-specific TS cells with the International Human Epigenetic Consortium (IHEC) and continue international contributions. 4) We establish a three-dimensional TS cell or TS-organoid culture system (an artificial placenta) using a channel device to recapitulate placental development and function in vitro. This system is useful to understand how the placenta responds to the intrauterine environment and will lead to innovative pharmaceutical products and contribute to health industry development. In summary, we will investigate the molecular mechanisms underlying human placental development and pregnancy-related diseases, which will contribute not only to the elucidation of the pathophysiology of pregnancy complications but also to the health of future generations.

Team Ushijima
Toshikazu Ushijima
President, Hoshi University
Research Content
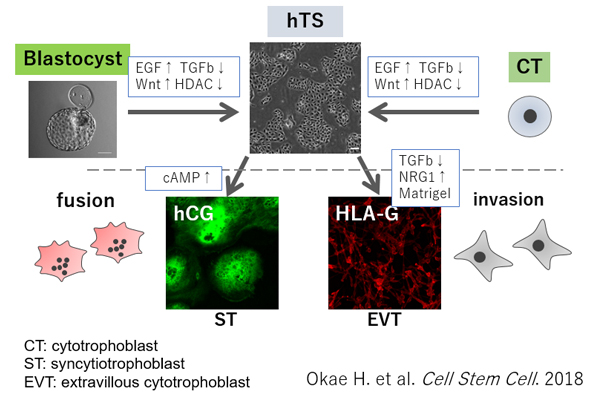
Cancers in adolescents and young adults (AYAs) are composed of three categories; embryonic tumors that developed in AYAs, tumors characteristic to AYAs, such as bone tumors, and cancers typical in the elderly but developed in AYAs. Gastric, uterine, and breast cancers, often found in the elderly, are more aggressive in AYAs than in the elderly. However, the reason remains largely unknown.
We have elucidated that chronic inflammation due to Helicobacter pylori infection causes abnormalities in the epigenome, resulting in the development of cancers such as gastric cancer. We have developed a method to determine the extent of the epigenomic abnormalities in gastric cells due to H. pylori infection, and discovered that the degree of abnormality is correlated with gastric cancer risk. When mice were infected with H. pylori, epigenomic abnormalities were induced similar to that in humans. The longer the infection period, the greater the degree of epigenomic aberration.
The epigenome is an important mechanism via which cells adapt to their environment. In childhood, adolescence, and young adulthood, the cellular epigenome is tuned up in response to various environmental factors. Therefore, the epigenome in cells from AYAs seems to have a high “plasticity,” or ability to adapt to the environmental stimuli. Indeed, the gastric cells of young mice showed higher epigenetic plasticity than those of adult mice. We hypothesized that this high plasticity also comes at a cost; epigenomic instability can occur when cells are exposed to carcinogens such as H. pylori infection.
Therefore, this research aims to show that this high plasticity in children and adolescence can also be linked to epigenomic instability under conditions of exposure to carcinogens. We will elucidate the genes involved in the maintenance of epigenomic plasticity, and identify the stimuli and genes involved in the conversion of plasticity to instability. We will prove the function of those genes using genetically engineered mice. Finally, we will establish a strategy to prevent highly malignant cancers in AYAs using the mouse models.
Contribution to IHEC
To identify genes involved in epigenomic plasticity, we will analyze several types of epigenomic modifications (DNA methylation, H3K27me3, H3K4me3, etc.) in multiple types of gastric cells (epithelial cells, fibroblasts, immunocytes, etc.) from juvenile and adult mice. Owing to the complexity of these analyses, we will collaborate with Dr. Ryuichiro Nakato in CREST-PRIME “Early Life.” The analysis method to be developed is expected to greatly contribute to the “Integrative Analysis” activity of IHEC. Simultaneously, the epigenomic data obtained from human cells will be registered to IHEC.
Project Team Leader
- Toshikazu Ushijima
President, Hoshi University
Topic Title: “Mechanism of Epigenomic Instability and Establishment of a Strategy to Prevent Aggressive Cancers in Adolescent and Young Adults”
Major Research Collaborator
- Hiroaki Honda
Director and Professor, Institute of Laboratory Animals, Tokyo Women's Medical University
Topic Title: “Engineered Animal Production and Analysis of Epigenomic Instability”

Team Nakato
Ryuichiro Nakato : Associate Professor, Laboratory of Computational Genomics, Institute of Quantitative Biosciences, The University of Tokyo
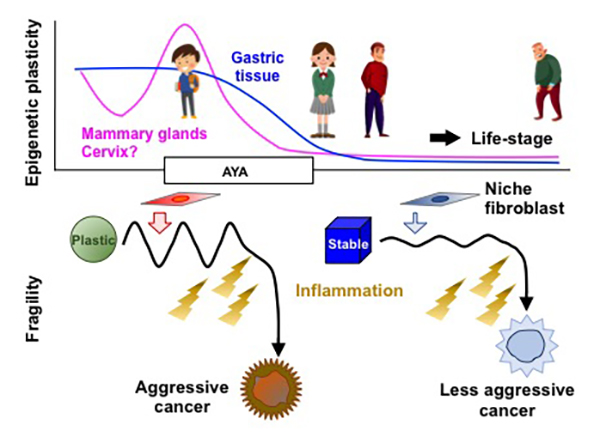
After completing the human genome by Human Genome Project in 2003, there has been an effort to identify and annotate the functional regions in the genome. Notably, the epigenomic information consisting of histone modifications and DNA methylation could reflect information about aging and life stress. This information could be inherited by the next generation, resulting in the risk of disease in offspring. Therefore, it is of importance to clarify the regulatory mechanisms and critical genomic regions. Our group is specializing in the computational analysis of genome and epigenome using next-generation sequencers (NGS) and has been developing new analysis methods to acquire new biological insights. We participated in the IHEC phase I as a member of the Shirahige group, a pioneer of the ChIP-seq field, from 2012–2016. In this project, our group cataloged gene expression and active histone marks in different types of human endothelial cells and performed a comprehensive analysis with chromatin interaction data to understand their diverse phenotypes and physiological functions.
In this AMED-IHEC project, we focus on "data-driven large-scale NGS analysis," a central topic in IHEC phase II, which achieves an epoch-making biological discovery from large NGS datasets (~hundreds of samples) without help from existing biological knowledge. Such a large-scale analysis has the potential to extract complex relationships and regulatory mechanisms in genomic and epigenomic information. The abundant dataset generated in the IHEC phase I can allow such an analysis. However, there is a substantial variance of data quality derived from biases and batch effects due to technical problems, because multiple laboratories were responsible for data acquisition using different tissues and protocols. This is problematic because data-driven approaches are sensitive to noise in the input dataset. To overcome this problem, we aim to develop a robust procedure for comparative epigenome analysis that circumvents variations at the level of the individual and technical noise derived from sample preparation under various conditions. This system will increase the value of existing epigenome databases and the epigenome field itself. As a representative of Japan in the field of epigenome information analysis, we progress data-driven NGS analysis by discussing with the world's leading experts and contribute to the further development of epigenome research.
 IHEC Committee & Working Group Members
IHEC Committee & Working Group Members
International Scientific Steering Committee
Toshikazu Ushijima
President, Hoshi University
Hiroyuki Sasaki
Distinguished Professor, Division of Epigenomics and Development, Medical Institute of Bioregulation, Kyushu University
Hiroyuki Takeda
Professor, Department of Biological Sciences, University of Tokyo
Assay Standards & Data Ecosystem
Hiroshi Kimura
Professor, Cell Biology Center, Institute of Innovative Research, Tokyo Institute of Technology
Mikita Suyama
Professor, Division of Bioinformatics, Medical Institute of Bioregulation, Kyushu University
Bioethics
Yae Kanai
Professor, Department of Pathology, Keio University School of Medicine
Integrative Analysis
Ryuichiro Nakato
Associate Professor, Laboratory of Computational Genomics, Institute of Quantitative Biosciences, The University of Tokyo
Mikita Suyama
Professor, Division of Bioinformatics, Medical Institute of Bioregulation, Kyushu University