





 Post-bisulfite adaptor-tagging (PBAT)法
Post-bisulfite adaptor-tagging (PBAT)法
三浦史仁、伊藤隆司
九州大学大学院医学研究院 医化学分野
 全ゲノムバイサルファイトシーケンシング(WGBS)
全ゲノムバイサルファイトシーケンシング(WGBS)
DNAのメチル化(シトシンの5位炭素のメチル化)は重要なエピジェネティック機構で、細胞分化・胚発生・様々な疾患など多くの生命現象に深く関わっています。ゲノムDNAのメチル化パターンは細胞種ごとに異なると考えられており、わずか数塩基のメチル化の違いが特異的な表現型の差異を惹き起こす例も知られています。したがって、出来るだけ多くの種類の細胞について、ゲノム上の全シトシンのメチル化状態(メチローム)を調べることは、それぞれの細胞の性質を理解する上での重要な基盤になると期待されています。
現在、メチロームを最も詳細に把握できる技術は、全ゲノムバイサルファイトシークエンシング(WGBS)です。バイサルファイトという試薬でDNAを処理すると、シトシンはウラシルへと変換されます。一方、メチル化されたシトシンはこの変換を受けません(図1)。したがって、バイサルファイト処理を施したDNAの配列を決定して元々の配列と比較すると、個々のシトシンについてそのメチル化状態を知ることができます(図2)。この方法をバイサルファイトシークエンシング(BS)と呼びます。WGBSは、バイサルファイト処理を施したゲノムDNA全体を次世代シークエンサーを用いて配列決定する方法です。IHECは、メチローム解析の標準手法としてWGBSを採用しており、様々な細胞や組織についてWGBSが試みられています。
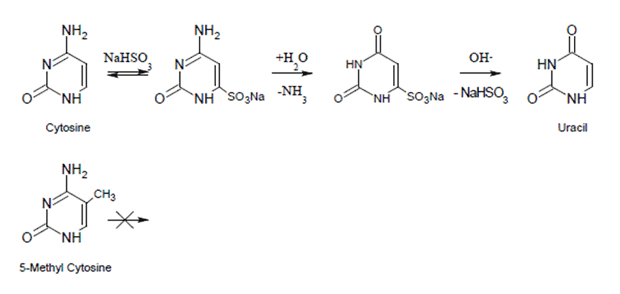
図1 バイサルファイト変換
バイサルファイト処理によってメチル化されていないシトシンがウラシルへ変換されるのに対して、メチル化されたシトシン(5メチルシトシン)はこの変換を受けない。(上)バイサルファイト処理によって、シトシンにはスルホン基が導入される。スルホン化されたシトシンは加水的な脱アミノ反応を起こして、スルホン化ウラシルへと変換される。アルカリ処理でスルホン基を除去するとウラシルが得られる。(下)5メチルシトシンはこの変換反応を受けにくい。
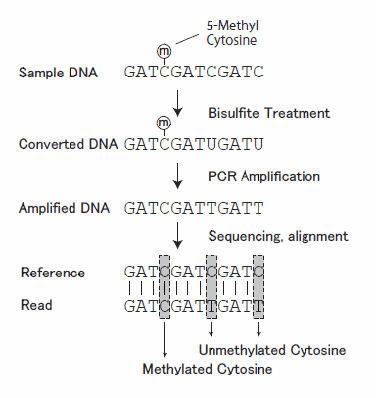
図2 バイサルファイトシークエンシング(BS)の原理
バイサルファイト処理によってDNA中のシトシン残基は、メチル化の有無に応じて5-メチルシトシンとウラシルに変換される。これらの塩基は、DNAポリメラーゼによってそれぞれシトシンとチミンとして複製されることになる。したがって、その配列(リード)を元来の配列(リファレンス)と比較すると、メチル化シトシンと非メチル化シトシンの部位は、それぞれC-CマッチとC-Tミスマッチとして検出される。
WGBSの問題点
バイサルファイト処理は、腐蝕性の化学物質であるバイサルファイト(亜硫酸水素)塩とDNAを混合して50~70℃に加熱する操作を含みます。DNAはこの過程で切断されてしまい、収率も極めて低いことが知られていました。最近になって収率のよい方法が開発されていますが、断片化を完全に回避することは出来ていません。
2008年、2つのグループが、モデル植物シロイヌナズナを対象に、WGBSに初めて成功しました。その際に一方のグループが用いたプロトコールMethylC-Seqは、その後の多くの研究で用いられており、IHECでも標準手法として推奨されています。しかしながら、MethylC-Seqでは、5 μgのDNAとPCRによる増幅が必要とされます。ヒト細胞は1細胞当たり6 pgのDNAを含みますから、MethylC-Seqを行うにはほぼ100万個(10の6乗個)の細胞が必要になります。培養細胞や均一な固形組織であれば、これだけの数の細胞を集めることは可能ですが、細胞種によってはそれが困難な場合が少なくありません。例えば、初期発生のメチロームを調べたくても、受精卵を百万個も集めることは不可能です。臓器を対象とする解析でもそれを構成する様々な細胞種をきちんと選り分けて解析しようとすると、百万個の細胞収集が現実的でなくなる場合が少なくありません。ヒトのあらゆる細胞や組織のエピゲノム解析を目指すIHECにとって、サンプル量の問題は、早晩、大きな壁となるでしょう。
PCRによる増幅は、少ないDNA量を補う上で有効ではありますが、バイアスを生むという危険性を孕んでいます。それぞれのDNA断片は、配列や塩基組成の違いによって、PCRで増幅される効率が異なります。したがって、PCRのサイクル数が増えれば増えるほど、増幅されやすい配列とされにくい配列の間でその量に大きな差がついてしまいます。その結果、ゲノムのある領域は過剰に何回も読まれているのに対して、別の領域は全く読まれないようなことがよく起こります。さらにWGBSの場合、同じ配列でもメチル化状態の違いによってバイサルファイト変換後の配列には違いが生じてしまい、増幅効率にも差が生じます。その差がPCRで増強されてしまうと、肝心のメチル化率の推定を大きく誤ることにもなりかねません。特に量が少ないサンプルの場合には、PCRのサイクル数を増やさざるを得ず、この危険性が非常に高まります。
このような問題点を考慮すると、理想的なWGBS法とは、MethylC-Seqよりもぐっと少ない量のDNAからスタートできて、可能ならばPCRによる増幅を含まない方法です。そんな方法があれば、メチローム解析の対象は今よりも格段に拡がり、得られるデータの質もより良質のものになるでしょう。
PBATによるWGBSの革新
私達は理想的なWGBS法を開発するために、従来法が大量のDNAを用いながらPCR増幅を必要とするのはなぜかについて検討を行いました。その結果、ゲノムDNA断片にアダプターを付加した後にバイサルファイト処理を施すことが、収量低下の大きな原因であることに気が付きました。次世代シークエンサーで配列決定を行うには、対象となるDNA断片の両端にアダプターと呼ばれる人工配列を付加する必要があります。しかし、両端にアダプター配列を付加したDNA(鋳型)をバイサルファイトで処理するとDNAの切断が起こってしまい、片側にしかアダプターが付いていない形、つまりシークエンスできない構造になってしまいます(図3)。つまり、バイサルファイト処理における収量低下もさることながら、切断によって鋳型として機能するために必須の構造が失われてしまうことが、従来法の鋳型調製効率が低い主な原因なのです。この効果は、DNAのサイズスタンダードをバイサルファイト処理してみると実感できます。ラダー状を示していたサイズスタンダードがスメア状に変化する有様は、バイサルファイト処理の過程でDNAに高頻度かつランダムに切断が導入されることを示しています(図4)。
では、この切断による鋳型構造の破壊を避けるには、どうしたらよいでしょうか?バイサルファイト処理後にアダプター付加を行えばよい筈です。そうすれば、アダプターが両端に付加されたDNAは、切断されることなく配列決定の際の鋳型として利用されるからです(図5)。これがPost-bisulfite Adaptor Tagging(PBAT)のコンセプトです。
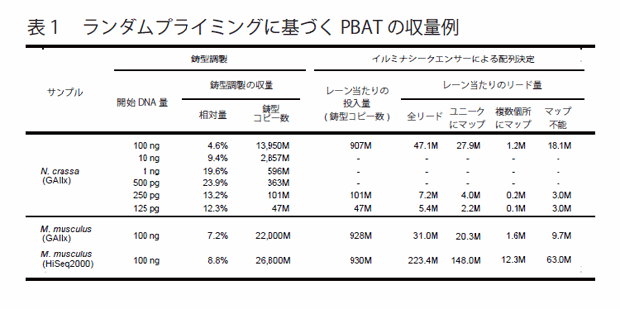
PBATを実現するために、我々は2回のランダムプライマー伸長反応によって鋳型を調製するプロトコールを開発しました(図6)。この簡便な方法は、従来法に比べて格段に効率的で、僅か125 pgのDNAからでもPCR増幅なしに鋳型を調製することが可能なほどでした(表1)。実際、PBATはマウス卵細胞1000個からのPCRフリー解析等の極微量サンプルの解析で大いに活躍しています。5000細胞に相当する30 ngのDNAがあれば、IHECで求められる量のデータをPCR増幅なしに産生することも可能です。
このようにPBAT法は、メチローム解析の対象の幅を拡げ、データの質を高めてくれることから、IHECにおける強力な武器になると期待されます。また、PBATはランダムプライマー伸長法以外でも実現可能でしょう。それらの可能性も含めて、我々はPBAT法の更なる高度化を進めています。
PBAT法の最新プロトコールは以下のリンクから手に入れることが可能です。
![]() Post-bisulfite adaptor tagging プロトコールリビジョン 12
Post-bisulfite adaptor tagging プロトコールリビジョン 12
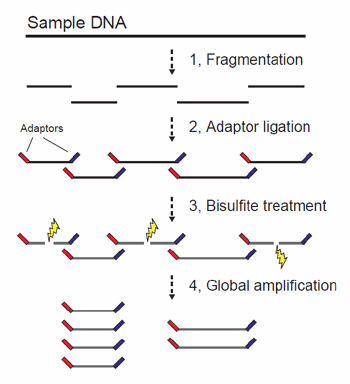
図3 バイサルファイト処理における鋳型構造の破壊が従来型プロトコールの低収量の原因である
従来のWGBSプロトコールでは、ゲノムDNA断片の両端にアダプター付加した後で、バイサルファイト処理を行っていた。バイサルファイト処理に伴うDNAの切断によって、両端にアダプターを保持した構造が破壊されると、シークエンスの鋳型としては機能できなくなる。こうして破壊された鋳型分子はPCR増幅しても再生されることはない。
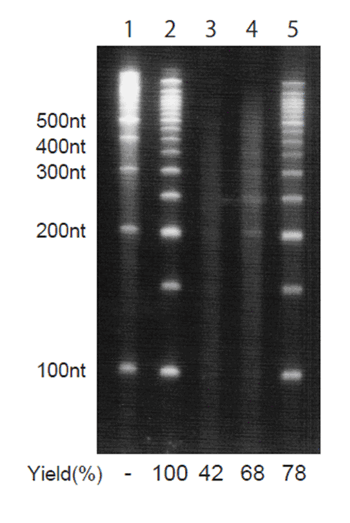
図4 バイサルファイト処理におけるDNAの分解
市販のキットを用いてDNAサイズスタンダード(50-bpラダー)にバイサルファイト処理を施し、蛍光色素による定量で収率を計算するとともに、変性ポリアクリルアミドゲル電気泳動を行った。レーン1:100-bpラダー。レーン2:インプットの50-bpラダー。レーン3:加熱変性後にバイサルファイト処理を行った50-bpラダー。レーン4:アルカリ変性後にバイサルファイト処理を行った50-bpラダー。レーン5:バイサルファイト処理以外の操作のみを行った対照。各レーンの下の数字は収率。
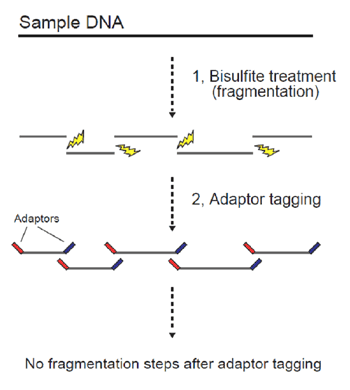
図5 Post-bisulfite adaptor tagging (PBAT)のコンセプト
バイサルファイト処理の後でアダプターを付加すれば、バイサルファイト処理における鋳型構造の破壊を防ぐことができる。
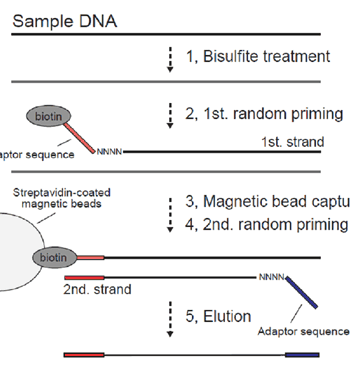
図6 ランダムプライミングに基づくPBAT
バイサルファイト処理を施したゲノムDNAを鋳型にして、ランダムプライマー伸長反応を2回行うと、両端にアダプターが付加された鋳型を効率的に調製することができる。
- Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 40, e136 (2012).
- Miura F, Ito T. Highly sensitive targeted methylome sequencing by post-bisulfite adaptor tagging. DNA Res. 22, 13-18 (2015).